Introduction
The brain receives and consumes 20% of all metabolic reserves of the body but it has a very limited capacity to store energy and so is very vulnerable to any focal or global event of hypoperfusion. The brain consumes oxygen at a high rate and so needs a reliable and constant supply. The brain only accounts for 1-2% of body mass but uses up 12-15% of cardiac output. Normal Cerebral blood flow (CBF) is about 50 ml per minute for every 100 g of brain mass. There is a close regulation of local blood flow to match needs called flow-metabolism coupling.
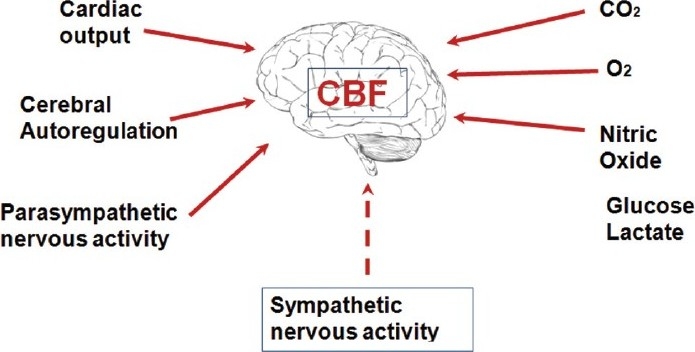
Cerebral blood flow is determined by several factors the most important of which are hyperaemia and autoregulation. Autoregulation ensures a controlled cerebral blood flow even with wide fluctuations of BP within the range of 50-160 mmHg. Hyperaemia may be seen in physiological situations, such as reading, calculating, or any other mental exercise. This neuronal-vascular responsive association is termed the neurovascular unit [2]. Vasodilation and increased flow may be due to local byproducts of metabolism (potassium, H+, lactate, ATP), glutamate, and nitric oxide. There may also be a role for Peptides (vasoactive peptide, substance P, and others) exert effects on the nerves that innervate cerebral vessels. Neurogenic control of CBF occurs by sympathetic innervation and is independent of the influence of PaCO2.
The fundamental physiological building block of the CNS is the neurovascular unit. This is composed of neurones, interneurones, astrocytes, basal lamina covered with smooth muscular cells and pericytes, endothelial cells and extracellular matrix. Astrocytes play an active role in coordinating blood flow as they communicate simultaneously with both neurones and blood vessels.
Cerebral Blood flow Autoregulation
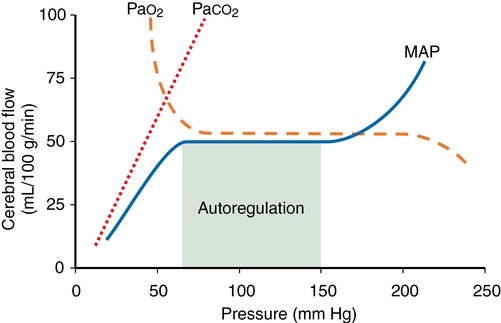
Autoregulation is defined as the maintenance of CBF over a range of mean arterial pressure (MAP) (see Figure 42-1). Cerebral vascular resistance controls CBF. Cerebral perfusion pressure equals MAP minus intracranial pressure (ICP). Because ICP (and therefore cerebral perfusion pressure) is not commonly available, MAP is used as a surrogate of cerebral perfusion pressure. Autoregulation occurs when MAP is between 70 and 150 mm Hg in the normal brain. This is a conservative estimate, given that considerable interindividual variation occurs. At the lower limit flow falls and at the upper limit flow increases.
Cerebral vascular resistance varies directly with BP to maintain flow. There is a momentary delay of 1-2 min for flow to adjust after an abrupt change in blood pressure. In hypertensive patients this curve is shifted tot he right and such a patient may be at risk for developing brain ischaemia at low MAP. Several weeks of blood pressure control may normalise the curve.
Basic causes of Ischaemic stroke
The basic causes of ischaemic stroke are thrombosis, embolism and atherosclerosis and systemic low flow. These causes may well overlap.
Cell Death in Ischaemic Stroke
If a sufficient injury occurs then cell death proceeds though a variety of steps often called a cascade. Cell death in stroke is due to one of at least two complex events, cytotoxic cell death and programmed cell death (apoptosis). The fundamental difference is that the first elicits an inflammatory response whereas the second doesn't. Mechanism include excitotoxicity, calcium overload and oxidative stress. The stages of cytotoxic cell death are shown below. Haemorrhagic strokes are not the one event and then subsequent damage. Brain perfusion can also be impaired due to damaged autoregulatory processes and then cerebral perfusion becomes very much dependent on systemic blood pressure - which can have consequences if this is too high or too low.
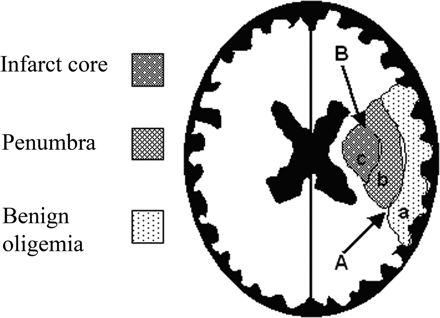
Cerebral blood flow (CBF) reduction below critical thresholds discriminates between irreversible infarct core, penumbra, and benign oligaemia (penumbra that recovers spontaneously). Thresholds are based on animal studies, and their diagnostic accuracy in humans has never been established. The optimal reported CBF thresholds varied widely, from 14.1 to 35.0 and from 4.8 to 8.4 mL/100 g per minute for penumbra and infarct core, respectively. Grey matter uses 2.5 times more ATP than white matter [1]. The penumbra is the most clinically relevant target for thrombolysis and thrombectomy and reperfusion therapies. Rate of progression to infarction depends on the degree of collateral arterial circulation, duration of insult, and functional and metabolic cellular state [1]. Much work is focused on trying to identify those patients with large penumbra and small infarct core who may benefit from reperfusion strategies beyond the current thrombolysis and thrombectomy windows.
Stages of Hypoxic Injury and Death
A lack of perfusion soon leads to a failure of cellular metabolism generating insufficient ATP. This leads to a a failure of membrane pump activity, failure to depolarise and a failure of the Na+ and K+ pumps, leading to depolarization of the neuronal membrane. Depolarization causes neurons to release glutamate into the synaptic cleft. Glutamate release increases cellular excitability and ATP demand and causes excitotoxicity [Castillo J et al. 1996]. Glutamate is the most common excitatory neurotransmitter. Glutamate neurostimulation leads to the opening of calcium channels associated with N-methy1-D-asapartate (NMDA) and alpha-amino-3-hydroxy-5-methyl-4-isoxanole propionate (AMPA) receptors. Persistent membrane depolarisation causes influx of calcium, sodium, and chloride ions and efflux of potassium ions. A rise in intracellular calcium activates proteases, lipases and endonucleases with the release of cytokines and causes further cellular damage. Mitochondrial damage can lead to apoptosis and iNOS (inducible Nitric oxide synthase) release and the formation of free radicals. There is breakdown of phospholipase and ultimately cytotoxic cell death. Blood-brain barrier integrity is reduced [Brown RC et al. 2002]. The additional paradox is that with the abundance of free radicals and other chemical species, reperfusion of these areas is preferable but is also not without its potential downside [Kuroda et al 1997].
Ischaemic Stroke
Artery to Artery Embolic Stroke
The main source of embolism in total is from the valves and chambers of the left side of the heart due to inherent structural defects and underlying atrial fibrillation. Cardioembolic stroke is dealt with in this location. The other main cause is embolism from lesions within the arteries to occlude more distal smaller calibre arteries with thrombus.
The main cause of this is embolism comes from aortic or subclavian or internal carotid atheroma. Another important cause is thrombus forming over areas of arterial dissection. Aortic atheroma is detectable by TOE is significantly more common in stroke patients than health controls. Atheromatous plaques may be a source of embolic material through rupture and thrombus formation.
Clinical
The presentation may be sudden with an acute carotid or intracranial vessel occlusion and corresponding TIA or stroke neurology. A carotid bruit may be heard if there is carotid disease. Patient may have coexisting coronary artery disease, be a smoker or have hyperlipidaemia or hypertension.
| Cause | About |
|---|
| Carotid Atherosclerotic disease | Usually rupture of an unstable atherosclerotic plaque leads to overlying platelets activation and clot formation which embolises |
| Aortic Atherosclerotic disease | Usually rupture of an unstable atherosclerotic plaque which releases debris into the carotids/vertebral |
| Subclavian Atherosclerotic disease | Usually rupture of an unstable atherosclerotic plaque which releases debris into the vertebral |
| Carotid Artery Dissection | Usually rupture of the intima with overlying platelets activation and clot formation which embolises |
| Vertebral Artery Dissection | Usually rupture of the intima with overlying platelets activation and clot formation which embolises |
| Intracranial Artery Atherosclerotic | Commoner in Afro-Caribbean. Can be plaque rupture and embolisation |
Management: If there is > 50% carotid stenosis which is symptomatic and the likely source of emboli then carotid endarterectomy (or stenting) should be considered. Antiplatelets are started and in some cases dual antiplatelets are sometimes considered. Smoking cessation is key. Aggressive lipid lowering, blood pressure control and management of vascular risk factors is advocated. Finally, most embolism is thrombotic but other rare sources of emboli are air, fat, cholesterol, bacteria, tumour cells, and particulate matter from injected drugs should be considered in rare cases.
 Carotid plaque
Carotid plaque
Cardioembolism
Cardioembolism refers to strokes due to emboli emanating from the heart. There are several underlying reasons why this can happen. The high flow rate of blood to the brain means that it is inherently susceptible to cardioembolism. Cardioembolic strokes related to AF and PAF are very preventable with anticoagulation and so detecting AF/PAF is incredibly important. There are other less common aetiologies. Cardioembolism accounts for 20% of ischaemic strokes. Cardioembolic strokes are generally more severe and are more prone to recurrence than other stroke types. The risk of long term recurrence and mortality are high after a cardioembolic stroke. By far the most common cause of cardioembolic stroke disease is non valvular Atrial fibrillation.
Cardiac emboli tend to flow to the intracranial vessels. Over two-thirds of blood goes via the anterior circulation and most of the time thrombi occlude the proximal MCA causing often massive, superficial or single large striatocapsular or multiple MCA territory infarcts. Clinical effects will depend on the proximity of occlusion in the MCA, pre-existing collateralisation, rapidly or reperfusion of the vessel, thrombus break up and distal embolisation and thrombolysis. Be aware that a large clot or a pre-existing carotid stenosis can cause thrombus to cause acute internal carotid artery subacute or total occlusion. Posterior circulation emboli can occlude vertebral and basilar arteries as well as reaching distal posterior cerebral artery. Brainstem, cerebellar and occipital lesions can be seen which may be multiple.
Classical teaching is that embolic strokes are clinically maximal at onset. A shower of emboli or emboli breakup and distal carriage can cause simultaneous multi territory strokes. Coma may be seen with rapid improvement. It is also recorded that embolic strokes can show sudden improvement. Recanalisation and haemorrhagic transformation also commoner with embolism. Lacunar strokes especially multiple are less likely to be cardioembolic and tend to be more likely small vessel related.
Brain imaging is useful but until there are bilateral or anterior and posterior circulation large vessel stroke lesions the CT does not help greatly with aetiology. MRI in this respect can show more detail, can identify older lesions not seen on CT and show that subcortical strokes extend to cortex and more likely to be large vessel. CT may show Haemorrhagic transformation seen in up to 71% of cardioembolic strokes [Jörgensen L et al. 1969]. Haemorrhagic infracts are predominately cardioembolic [Fisher CM et al. 1951].
Making a diagnosis of Cardioembolism is usually supported by finding of either AF or a structural cardiac lesion. The history of examination findings of AF or rheumatic valve disease or prosthetic valves makes the diagnosis straightforward. Difficulties arise when AF is paroxysmal but in acute stroke it should be detectable acutely and if not present then Holter monitoring should be considered really for at least 48 hours. There should be a very low threshold for looking for PAF or sick sinus syndrome both of which are associated with embolism. Transthoracic echocardiogram can detect the typical findings of mitral stenosis or dilated cardiomyopathy and other structural ventricular diseases. Left ventricular thrombus, valve vegetations, or atrial myxomas can also be detected. Left atrial size and left ventricular systolic function can be assessed. In younger patients or when valve or other structural lesions need better defined then Transoesophageal echocardiogram is used. Diagnostic views of aortic arch, ascending aorta, left atrium and left atrial appendages, intra-atrial septum, pulmonary veins, and valve vegetations can be seen. Any PFO or atrial septal aneurysm can be detected and bubble contrast may be given to look for shunting.
Echocardiography in the first hours after the onset of stroke is necessary only in rare cases, such as if infective endocarditis is suspected. In the days thereafter, transthoracic echocardiography or, preferably, transoesophageal echocardiography can be undertaken to support or refute a possibility of cardioembolism.
Causes and Risk of Cardioembolic stroke [Tudor G. et al. 2008]
| High Risk | Low or uncertain Risk |
|---|
| Atrial Fibrillation | Patient Foramen Ovale |
| Sustained Atrial Flutter | Atrial septal aneurysm |
| Sick Sinus Syndrome | Spontaneous atrial contrast |
| Left Atrial Thrombus | Mitral Valve prolapse |
| Left Atrial Appendage Thrombus | Calcific aortic stenosis |
| Left Atrial Myxoma | Fibroelastoma |
| Mitral Stenosis | Giant Lambel Excrescence |
| Mechanical Valve | Akinetic Ventricular wall segment |
| Infective Endocarditis | Subaortic hypertrepophic cardiomyopathy |
| Non Infective Endocarditis | Congestive cardiac failure |
| Left Ventricular Myxoma | |
| Recent Anterior MI | |
| Dilated Cardiomyopathy | |
Structural Heart Disease as a source of Cardioembolism
These include any condition that allows stasis and the formation of thrombi. Severely Impaired LV function, LV aneurysm formation. Atrial or Ventricular myxomas. There may be coexisting AF. Others include Cardiomyopathy, Mitral valve prolapse and prosthetic heart valves.
| Cause | About |
|---|
| Cardiomyopathy | Risks of cardioembolism may be seen in those with very poor LV function and those with underlying Chronic AF with enlarged LA. |
| Mitral Valve Prolapse | Studies have shown that mitral valve prolapse (MVP) is associated with fibrinous deposits on the valve, endothelial denudation and annular thrombus at the junction with the atrial wall. The myxomatous redundant valve leaflets appear to increase the predisposition to thromboembolic events. There is no evidence for antithrombotic treatment other than antiplatelets in those who have experienced stroke. There should be a search for undetected PAF. |
| Mitral annulus calcification | Is seen with mitral stenosis, mitral regurgitation and may be increased in those with cardiogenic brain embolism. Detected on echocardiography it suggests a twofold increase in risk for stroke. Embolism of fibrinated cell clot or calcium spicules has been reported. |
| Prosthetic Heart Valves | Diseased and damaged heart valves are often replaced with either mechanical or bioprosthetic (tissue) valves. Tissue prosthetic valves are believed to be associated with a smaller risk of thromboembolism than mechanical valves. Mitral valve prostheses are associated with a greater risk of thromboembolism than Aortic, possibly because of the higher incidence of atrial fibrillation and other thromboembolic risk factors in these patients. |
Paradoxical Embolism
The pulmonary circulation provides a large filter for any thrombi returning from the systemic circulation. A small microthrombus to the lungs is probably insignificant but if the pulmonary circulation can be bypassed and shunted then there is a risk of thrombi entering the systemic arterial circulation. This can happen when there are areas allowing right to left shunting in the heart such as a patent foramen ovale or Atrial septal defect or Ventricular septal defect or other form of congenital heart disease. However, clots bypassing the lungs can be seen in those with pulmonary arteriovenous malformations. To reliably diagnose paradoxical embolism, I would suggest you need to try to prove
- A stroke that is Radiologically cardioembolic - i.e. large artery and perhaps multiple territories
- Evidence of a DVT at the time - Doppler USS or at least a raised Dimer
- Evidence of a shunt with Right to left flow
- It helps if there is an underlying venous procoagulant state e.g. malignancy, pregnancy etc.
Management is difficult. First of all any PFO which is seen in 25% of the population may simply be a bystander and association does not mean causation. Closure of PFOs is not risk free and may even increase long term risk of (P)AF and so limits any reduction, cancels, or even increases any risk of stroke. Unfortunately, the interventionalist have not proven their case. All those having PFO closure should be done as part of a proper clinical trial.
Localised Atherosclerosis and vessel stenosis and occlusion
Atherosclerotic plaques are seen in postmortem studies of the aorta and other large and medium sized vessels of patients even from their 20's. These plaques can degenerate and rupture leading to thrombus in situ which may cause complete or subtotal occlusions, and which can embolise distally. This can be found in aortic or subclavian or internal carotid atheroma. Another important cause is thrombus forming over areas of arterial dissection where the intimal integrity is lost. Aortic atheroma is detectable by TOE is significantly more common in stroke patients than health controls. Atheromatous plaques may be a source of embolic material through rupture and thrombus formation.
Clinical
- Transient vessel occlusion and corresponding TIA
- Acute stroke. A carotid bruit may be heard.
- Coexisting coronary artery disease
- Smoker, diabetes or have hyperlipidaemia or hypertension.
Causes of Medium/Large Vessel Disease
| Cause | About |
|---|
| Atherosclerotic disease | Disease may be seen both in the large and medium-sized arteries from the aorta to carotids and subclavian and vertebral vessels as well as extracranial and intracranial vessels which are commoner in those of Afro-Caribbean origin. Lesions may be seen at typical sites such as bifurcation of the carotid (branching), aortic arch, carotid syphon (tortuous) and the basilar artery (confluence). Atheroma seems to accumulate at sites of haemodynamic sheer stress. Usually, rupture of an unstable atherosclerotic plaque leads to overlying platelets activation and clot formation which embolises. The first sign of the disease is the formation of fatty streaks and with time smooth muscle cells proliferate and there is local movement and accumulation of lipid. The plaques have a fibrous cap which can become unstable and fissure and crack and rupture with the release of prothrombotic matter. The plaques can also cause luminal obstruction and narrowing. Close relationship with age, male sex, smoking, cholesterol, hypertension, diabetes, obesity, lack of exercise
|
| Cervical Artery Dissection | Usually due to a tear of the intima with subintimal bleeding. There is luminal platelet activation over the area and clot formation which embolises. There may be local aneurysmal formation. Causes of dissection include local trauma and presumed mechanical damage or fibromuscular dysplasia. See dissection. |
| Fibromuscular dysplasia | Often presents with dissection type clinical appearance. Commoner in younger females. Affects small and medium-sized arteries. Can affect the mid portion of the ICA. Angiography may show a classical string of beads like appearance, and there can even be a web-like lesion. |
| Irradiation | Patients with prior direct irradiation of the neck and brain can go on later to suffer arterial strokes. The mechanism is unclear - a radiation vasculopathy or accelerated atherosclerosis |
| Moya Moya disease | There may be stenosis or occlusion of the distal ICAs unilaterally or bilaterally. Can have involvement of the circle of Willis and sometimes of the proximal cerebral and basilar arteries.
|
| Giant cell arteritis | Older patients. Tends to affect the posterior circulation preferentially though stroke is rare. |
| Vasculitis | May be a systemic or cerebral vasculitis. Localised inflammatory change and luminal changes and oedema can cause ischaemia/infarction. |
| Varicella vasculopathy | Can occur without rash. Localised inflammatory change and luminal changes and oedema can cause ischaemia/infarction. |
Artery to artery emboli
- Management: If there is > 50% carotid stenosis which is symptomatic and the likely source of emboli then carotid endarterectomy (or stenting) should be considered. Anti-platelets are started, and in some cases, dual antiplatelets are sometimes considered. Smoking cessation is key.
- Aggressive lipid-lowering, blood pressure control and management of vascular risk factors is advocated. Finally, most embolism is thrombotic but other rare sources of emboli are air, fat, cholesterol, bacteria, tumour cells, and particulate matter from injected drugs should be considered in rare cases.
Small vessel Disease
Small vessel disease accounts for about 25% of the causes of Ischaemic stroke. It is also the main underlying pathology for deep bleeds which are discussed in the chapter on haemorrhagic stroke. It is closely associated with Hypertension. Small vessel disease is as its name suggests a disease predominately of the small penetrating arterioles usually less than 800 um in diameter. They often are found branching off sideways from small and medium sized arteries which come off the circle of Willis or vertebrobasilar systems. They cannot be imaged in vivo so we use surrogate markers - namely lacunar strokes and deep haemorrhages as evidence of its presence. Important caveat is that small vessels disease can be due to large vessel disease such as proximal MCA embolic occlusion which may reperfuse after causing localised deep infarction or large plaques in larger vessel which can encroach on the ostia or branching small vessel causing occlusion. Multiple acute lacunar type infarcts may suggest embolism.
There may be some over exposure to high blood pressure as there is not the steady reduction in vessel calibre seen in other systems. The occlusion of these small vessels can lead to predominantly white matter damage and demyelination with axonal damage and scarring. In most cases a process called lipohyalinosis which seems different to atherosclerosis occludes the vessels. Other processes exits including amyloid and inherited genetic defects. An aetiological classification is shown below. The result is small deep hemispherical or brainstem (mainly pontine) strokes usually less than 15 mm in diameter. Hypertension and diabetes appear to be risk factors. This pattern of disease may also be seen with CADASIL, Fabry's disease, Vasculitis, migraine and other uncommon causes of stroke [Pantoni L 2010]. Don't forget that it is also disease of these small vessels that can lead to the classically deep haemorrhages that can be disabling or fatal.
Other rarer causes of small vessel strokes include localised radiotherapy, CADASIL, Fabry's disease. These will be discussed separately later.
Detailed classification of Small Vessel Disease [Pantoni L. 2010]
This classification and table need not need memorised but what is important is to realise that small vessel disease is multifactorial with a broad differential and is in reality a number of pathological processes all affecting the small penetrating vessels.
| Type and Title | Description |
|---|
| 1 : Arteriosclerosis | Commonest form. Pathology shows Fibrinoid necrosis, Lipohyalinolysis, Micro atheroma, Microaneurysms. Similar disease seen in kidney and retina. Associated with increasing age, diabetes and hypertension. |
| 2 : Sporadic hereditary amyloid | Beta A4 Amyloid deposition in walls of small and medium sized arteries and arterioles. May be blood extravasation. Associated with Alzheimer disease. Association with microbleeds on MRI. Unsure who gets microbleeds and others get large bleeds. |
| 3 : Inherited/Genetic Defects | CADASIL, CARASIL, MELAS, Fabry's disease, Hereditary cerebroretinal vasculopathy, multi-infarct dementia of Swedish type, COL4A1 mutations |
| 4: Inflammatory/Immune (Vasculitis) | Wegener's granulomatosis, Churg-Strauss, Microscopic polyangiitis, Henoch-Schönlein, Cryoglobulinaemic vasculitis, Sneddon's syndrome, SLE, RA, Sjogren's syndrome |
| 5 : Venous Collagenous | |
| 6 : other small vessel disease | Post radiation angiopathy |
Clinically the effects are often subclinical but slightly larger occlusions can lead to lacunar infarcts which can certainly present with the classical lacunar syndromes discussed elsewhere. A more piecemeal damage can lead to a progressive vascular dementia, Gait apraxia and vascular Parkinson's as deep white matter is affected as well as subcortical nuclei and their connections.
Imaging is useful and CT often shows hypodensity most distinctly around the ventricles with associated cerebral atrophy. MRI is much more sensitive and shows increased signal on T2 and FLAIR. It is a very similar appearance and distribution to demyelination. Resulting lacunar infarcts are usually round and less than 1.5 cm in diameter. Diffusion weighted imaging is useful to pick these up acutely.
With Small vessel disease other MRI with gradient echo may show evidence of microbleeds showing the two sided nature of this disease. Doppler and Echo have probably less importance but should be done where indicated and where there is diagnostic doubt if this was a small vessel stroke or cardioembolism with a shower of small emboli occluding vessels.
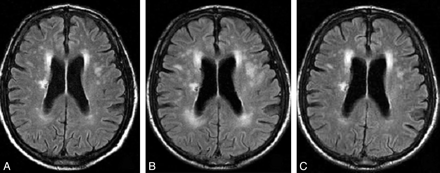
Small Vessel Disease concentrated typically around the ventricles
Management
General management of small vessel disease is to first see if there is any suggestion of any of the more unlikely causes and test if there is especially in those under the age of 50 (Fabry's disease, CADASIL). However most of the time it is old age, hypertension and diabetes that seem to be the main risks. Manage any hypertension, diabetes and glycaemic control and most patients would be started on an antiplatelet and receive lipid reduction and smoking advice. The SPS3 trial has shown that there is no benefit in giving dual antiplatelet therapy (aspirin and clopidogrel) rather than aspirin alone in these patents [Benavente O 2011]. In the setting of acute stroke, those with small vessel disease should be considered for thrombolysis. The exact connection between small vessel stroke and larger vessel strokes is not fully understood. Carotid disease should also be addressed in those with symptomatic carotid disease and small vessel disease.
Low flow
Many strokes are simply to systemic fall in blood pressure and cardiac out and the most vulnerable areas in the brain are those that like in the edge of ACA and MCA and MCA and PCA territories these are the so-call watershed areas. One can surmise that systemic hypotension plus a coexisting stenotic lesion can lead to a combined critical fall in flow with infarction and this may be a marker of co-existing ipsilateral stenotic disease. This may be seen particularly with carotid stenosis.
Venous thrombosis and infarction
A blocked cerebral vein will lead to back pressure, oedema and even haemorrhage and infarction.
| Macroscopic changes post infarct develop with time |
|---|
- Cytotoxic and vasogenic oedema initially
- Liquefactive necrosis with time
- CT after several days shows radiodensity equal to CSF
- CT will show loss of volume and 'ex vacuo dilatation
|
|
Macroscopic appearance of Hypoxic Injury and Death
| Stroke Size and Shape |
|---|
- Territorial Infarcts: Wedge shaped infarction to the cortex following occlusion of a branch of Major cerebral artery
- Subcortical infarction as vessel reperfuses or good anastomosis
- Lacunar infarcts due to occluded deep penetrating vessel are small and circular
- Border-zone: Infarcted area between 2 large vessels when blood pressure drops
- Across arterial zones: Venous infarction, vasculitis
|
|
Cell Death in Haemorrhagic Stroke
The mechanisms of cell death in ischaemic stroke are again due to hypoperfusion of focal areas of brain due to rupture of the vessel affected and there can also be further physical effects. There is evidence that haemorrhage can affect the neurovascular unit described above. As mentioned above the rigid box principle applies very much to haemorrhage. The Monro-Kellie hypothesis states that the sum of volumes of brain, CSF, and intracranial blood is constant. The implication being that any added volume to any of these will be additional and lead to a rise in Intracranial pressure. This hypothesis has substantial theoretical implications in increased intracranial pressure and in decreased CSF volume which may help to balance any other volume.
There are some mechanism by which the brain can compensate. These include shunting away CSF from the brain to spinal subarachnoid space 'cisterns' and the dura can increase absorption. Changes in blood volume by vasodilation and vasoconstriction can control cerebral blood flow and maintain at a constant during variations in systemic BP. Cerebral veins are valveless and have little supporting tissue and are susceptible to compression.
However at some point ICP will begin to rise inexorably and this will lead to pressure downwards onto the brainstem with coma and death. In terms of haemorrhage the volume of 60 mls appears to be critical in terms of haematoma volume. However there is much variability and older patients with reduced brain volume due to atrophy may well decompensate at a higher mass volume. Haemorrhage may also be surrounded by a degree of oedema which can exacerbate the situation.
| Invasively Measuring Pressures |
|---|
Intracerebral Volume = Brain volume + CSF volume + Blood volume + Mass volume
CPP=MAP-ICP
CPP:70-100 mmHg
Normal ICP: 5 - 15 mmHg.
Usual treatment goal ICP < 20 mmHg
|
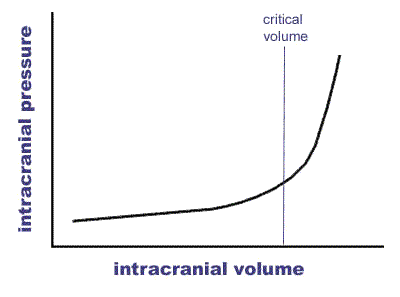
Rising Intracranial Pressure and Herniation syndromes
Cerebral oedema
Cerebral oedema increases brain volume. The most common form of oedema post stroke is cytotoxic oedema and this is due to breakdown of cell membrane function, influx of ions and water into the cell. It may be seen in the brain post stroke or global hypoperfusion e.g. cardiac arrest or with Reye's syndrome or some of the encephalopathies. Radiologically this shows as restricted diffusion on an MRI. The other form of oedema is vasogenic oedema and is primarily due to breakdown of the blood brain barrier which allows normal plasma constituents to enter the CSF. It may be seen in later stages of cerebral ischaemia and stroke and PRES (will meet this later). This shows as enhancement with contrast on a CT/MRI. It is characteristic of certain tumours and inflammatory processes. It is often managed with steroids. A third cause is osmotic oedema which is due to reduced plasma osmolality as is seen with severe hyponatraemia. Water enters the more hypertonic cells with a resultant increase in cellular volume and oedema. Death can result from raised ICP. Management is to slowly increase plasma osmolality. Elderly patients with pre-existing brain atrophy are often less vulnerable to pathological increases in brain volume as compared with young patients.
Managing Rising ICP
ICP is only measured directly in patients who get to a neurocritical ITU and this is a very small percentage, perhaps only really those who have required clot evacuation or aneurysmal surgery for SAH. Management is a balance between managing cerebral perfusion pressure and the intracranial pressure and ensuring there is adequate brain perfusion.
| Five ways to decrease ICP |
|---|
- Enhance venous drainage: Elevate head 30 degrees and ensure no neck constriction
- Hyperosmolar therapy e.g. Mannitol
- Hyperventilation - reduces carbon dioxide and causes vasoconstriction but is a short term strategy
- CSF Drainage : Neurosurgical Procedure - External ventricular drainage
- Surgery : Decompressive Hemicraniectomy +/- haematoma evacuation.
|
Raised ICP - avoid or minimise
| Avoid |
|---|
- Hypotension
- Hypoxia
- Hyponatraemia
- Hypervolaemia (avoid excess dextrose)
- Hyperglycaemia (manage using sliding scale initially)
- Hyperthermia (lower temp)
- Hypermetabolism (treat seizures and agitation)
|
Herniation Syndromes
| Herniation Syndromes |
|---|
| These are seen in the context of rising ICP in the comatose patient. These are all neurosurgical emergencies and demand rapid escalation. |
| Subfalcine | the brain is pushed under the falx separating right and left cortices. There may be compression and stroke involving the anterior cerebral artery with contralateral leg weakness. The lateral ventricles and brain midline will be distorted and shifted and can be seen on CT. |
| Transtentorial | A supratentorial mass or pressure gradient forces the uncus of the temporal lobe down beneath the tentorium 'tent' can result in an ipsilateral oculomotor paresis (dilated pupil, ptosis, down and out pupil) and compression of the posterior cerebral artery giving contralateral hemianopia. The shift forces the opposing cerebral peduncle against the edge of the tentorium causing contralateral hemiparesis to the peduncle. The indentation of the tentorium edge causes Kernohan's notch on the opposite cerebral peduncle. This is a false localising sign. |
| Transforaminal | the brainstem and cerebellar tonsils are forced down into the foramen magnum. Pressure on the medulla leads to apnoea and death |
References
- 1. Bandera E et al. Cerebral Blood Flow Threshold of Ischemic Penumbra and Infarct Core in Acute Ischemic Stroke
A Systematic Review. Stroke. 2006;37:1334-1339
- 2. Muoio V et al. The neurovascular unit. Acta Physiol 2014, 210, 790-798 ·